Nervceller som styr ögats muskulatur är ovanligt motståndskraftiga mot ALS.
Bild: Getty images
Tåliga celler ger hopp vid ALS
De flesta som får diagnosen ALS dör inom fem år. Men forskning på extra tåliga nervceller ger hopp om nya behandlingar, skriver Eva Hedlund som är professor i neurokemi.
Francis Tsai, en begåvad konstnär i USA, var 42 år gammal när han fick en domnad känsla i armarna. Han diagnostiserades med ALS (amyotrofisk lateral skleros), en sjukdom där nervceller som kontrollerar viljestyrda rörelser bryts ner. Det leder till muskelsvaghet och sedan förlamning tills andningen slutar fungera.
Francis Tsai blev snabbt sämre. Drygt ett år efter att han fått sin diagnos höll han i en penna för sista gången. Men det hindrade honom inte från att fortsätta med sin konst, som bland annat bestod av bilder på futuristiska rymdbosättningar i dunkla färger och miljöer till dataspel. När armarna slutade fungera målade han på en pekskärm med tårna. Och när det inte längre gick började han att rita genom att styra en dator med ögonen.
Francis Tsai målade – trots ALS
Ögats muskulatur drivs av nervceller som är exceptionellt motståndskraftiga mot sjukdomen. De fungerar även i slutstadier av ALS och används för att hjälpa patienter som inte längre kan tala att kommunicera med vårdpersonal och anhöriga. Tack vare sina bevarade ögonrörelser kunde Francis Tsai fortsätta med sin konst ända fram till sin död, fem år efter de första symtomen.
Kort livstid efter ALS-diagnos
I dag går det inte att bota ALS, men det finns läkemedel som kan bromsa förloppet något. De flesta som drabbas är mellan 40 och 70 år gamla, och patienterna lever vanligtvis mellan ett och fem år efter att de fått sin diagnos.
Jag började forska om molekylära mekanismer bakom ALS för mer än 20 år sedan. En viktig del av min forskning handlar om att ta reda på varför vissa nervceller är mer motståndskraftiga än andra. Målet är att identifiera vad som gör dem extra tåliga, för att sedan utveckla behandlingar som ger andra celler liknande motståndskraft vid ALS.
ALS kapar kopplingen mellan hjärnan och musklerna
- Amyotropisk lateralskleros, ALS, är en neurologisk sjukdom där nervceller i hjärnan, hjärnstammen och ryggmärgen bryts ner och dör. De nerver som drabbas kopplar ihop nervsystemet med kroppens muskler och kallas motorneuron.
- Musklerna förtvinar när motorneuronen dör och den som drabbas får allt svårare att röra sig, tala, svälja och andas. Processen går olika fort, men för de flesta leder ALS till döden inom några år.
- Även om främst motorneuron drabbas, får många patienter även kognitiv påverkan.
Sjukdomen är ovanlig. I Sverige insjuknar cirka 250 människor varje år. Men om man tittar på risken att utveckla ALS under en livstid så känns sjukdomen inte så ovanlig, risken att drabbas är cirka 1/300. Det är något jag ofta tänker på när jag går in i ett flygplan. Statistiskt sett kommer en av oss att drabbas av ALS någon gång i livet, och det känns skrämmande.
Sjukdomsförloppet är komplicerat. De nervceller som påverkas – så kallade motorneuroner – är stora och energikrävande och har sinsemellan olika egenskaper. De som styr ögonens rörelser finns i mitthjärnan och är speciella eftersom ögonen alltid rör sig, till och med under sömnen. Övriga motorneuroner finns längre ner i hjärnstammen och längs hela ryggmärgen. Deras utskott – i vissa fall över en meter långa – styr musklerna i hela kroppen ända ut i tå- och fingerspetsarna.
När motorneuroner dör blir musklerna svagare och förtvinar, vilket till slut leder till förlamning. Allra först drabbas motorneuroner som styr de snabba muskler vi behöver för att springa fort och hoppa långt. De tröttas ut fort och används bara korta stunder åt gången. Om man inte är professionell idrottare använder man dem bara några minuter per dag.
I ryggmärgen finns även långsamma motorneuroner som bland annat kontrollerar de muskler vi använder för att hålla kroppen i upprätt position, och även för långdistanslöpning. De är mer motståndskraftiga mot sjukdomen än de snabba, och har dessutom en viss förmåga att kompensera för sjukdomens skadeverkningar.
Fakta: ALS
- Så många drabbas av ALS: Mellan 250 och 400 svenskar varje år.
- Så många drabbas av SOD1: 2–5 procent av ALS-gruppen.
- Riskfaktorer: Hög ålder, manligt kön och ärftlighet.
- Första symtom: Ofta svaghet i ena handen eller foten. Ibland är första symtom talpåverkan.
- Vad händer: Nervcellerna i hjärna och ryggmärg bryts ned. Musklerna förtvinar.
- Vanligaste dödsorsak: Andningsmusklerna orkar inte längre.
Maja Lundbäck.
Källa: Karin Forsberg, neurolog och forskare vid ALS-forskningscentret vid Umeå universitet.
När snabba motorneuroner dör kan deras långsamma grannar delvis ersätta deras funktion genom att bilda nya kontakter med muskelceller. Detta leder samtidigt till att snabba muskelceller omvandlas till långsamma. På så vis kan sjukdomen stabiliseras under en tid. Men i takt med att även långsamma motorneuroner förstörs blir symtomen svårare.
Vad orsakar ALS?
I mitt laboratorium undersöker vi aktiviteten hos olika gener för att ta reda på varför vissa celler dör medan andra klarar sig under sjukdomsförloppet. Bland annat utvecklar vi olika metoder för att studera budbärarmolekyler för genetisk information (m-rna, som bildas av aktiva gener och styr cellernas tillverkning av proteiner). På så vis kan vi förstå hur en cell fungerar, hur den mår och på vilket sätt den hanterar den uppkomna situationen.
Genom sådana analyser har min och andras forskargrupper identifierat faktorer som antingen kan tillföras eller tas bort från nervceller för att öka deras motståndskraft. Vi har bland annat upptäckt att de motståndskraftiga motorneuronen som styr ögats muskulatur har ett antal unika faktorer, till exempel olika tillväxtfaktorer och synaptiska proteiner som verkar nervskyddande. Vi hoppas att detta i framtiden ska leda till nya behandlingar.
Bild: Melanie Leboeuf
En viktig fråga är förstås vad som orsakar ALS, hur kaskaden av skador på motorneuronerna börjar.
Riskfaktorer för ALS
Det finns ett antal tydliga riskfaktorer, inklusive ärftlighet och stigande ålder. Sjukdomen är också något vanligare bland män än bland kvinnor, åtminstone hos dem som insjuknar före 80 års ålder. I mellan 10 och 15 procent av alla fall av ALS finns en uppenbar ärftlighet.
När jag började forska om ALS kände man bara till en enda gen som i muterad form orsakar sjukdomen. Den genen kallas SOD1. I dag har man identifierat mutationer i mer än 40 olika gener som ökar risken att få ALS. Fyra av generna står för 60 procent av alla fall med tydlig ärftlighet i den europeiska befolkningen. (I Asien står de för 40 procent.) Mutationerna är nästan alltid är dominanta, vilket betyder att det räcker med en muterad kopia av genen från en förälder för att sjukdomen ska bryta ut.
Men i de allra flesta fall (85 till 90 procent) uppstår ALS sporadiskt – vilket betyder att vi inte förstår den exakta orsaken till sjukdomen, och att den inte heller verkar finnas i familjen sedan tidigare. Förutom genmutationer är som sagt stigande ålder den absolut viktigaste riskfaktorn.
Förut var det en vanlig uppfattning att dessa sporadiska fall orsakades av okända miljöfaktorer. I dag vet vi att bland annat rökning, miljögifter och kraftigt våld mot huvudet och ryggmärgen är riskfaktorer.
Samtidigt har det visat sig att genetik spelar en viktig roll även i de sporadiskt uppkomna fallen. Detta är påvisat genom tvillingstudier. Forskning har också visat att det finns en ökad risk att få ALS om man har en nära släkting som drabbats av sjukdomen sporadiskt. Risken ökar då från 0,3 procent (1/300 över en livstid) till 5,6 procent. Här är den bakomliggande genetiken komplex och vi förstår den ännu inte, men mycket talar för att flera faktorer kan vara inblandade.
I dagsläget handlar mycket forskning om de dominanta genmutationerna som orsakar ALS. Anledningen till att man fokuserar på att studera dem – och inte den sporadiskt uppkomna sjukdomen som är så mycket vanligare – är att man inte vet hur mycket sporadiska fall skiljer sig från varandra och det blir då mycket svårare att undersöka.
Olika typer av ALS
Det verkar som om ALS egentligen är ett samlingsnamn för många olika sjukdomar som alla leder till att motorneuroner dör. Därför är det förmodligen mer informativt att studera en specifik genmutation och undersöka hur celler reagerar på den. Sedan kan man gå vidare och undersöka om en annan genmutation ger samma respons, och hoppas att kunskapen även gäller för sporadiskt uppkommen sjukdom.
I mitt laboratorium har vi med detta arbetssätt kommit fram till att alla studerade genmutationer hittills påverkar cellernas energifabriker, mitokondrierna. Tillsammans med forskare vid Kings College London upptäckte vi nyligen att ALS påverkar motorneuronernas mitokondrier redan innan cellerna uppvisar andra sjukdomstecken, något som tidigare inte varit känt. Problem uppstår även när mitokondrier ska transporteras ut i neuronernas långa utskott där det finns ett stort behov av den energi de producerar.
Störningar i mitokondriernas funktion verkar alltså vara en gemensam nämnare för olika genmutationer som alla orsakar ALS. Den kunskapen öppnar för möjligheter att utveckla läkemedel mot sjukdomen oavsett vad som orsakat den.
En annan sak som är gemensam hos nästan alla patienter med ALS är att ett protein (TDP-43) ansamlas på fel ställe i cellerna. Den kunskapen kan leda till behandlingar mot sporadiska fall av ALS.
När det gäller tydligt nedärvda genmutationer börjar forskning leda till hopp om nya behandlingar för dessa specifika mutationer. Ett intressant exempel gäller mutationer i SOD1, den gen som först kopplades till ALS.
Genterapi mot ALS
Ett slags genterapi har visat god effekt på vissa patienter, vilket Forskning & Framsteg tidigare berättat om. En svensk man i 30-årsåldern som fick behandlingen inom en forskningsstudie kunde fyra år senare tala, gå i trappor och leva ett aktivt liv.
Mutationer i SOD1-genen gör att ett protein blir felveckat och klumpar ihop sig. Genterapin hindrar det felaktiga proteinet från att bildas genom att blockera budbärarmolekylen mrna som annars hade instruerat cellen att bilda proteinet (grafik). Själva läkemedlet heter tofersen och godkändes inom EU 2024 under namnet Qalsody. Det ges i form av en spruta var fjärde vecka in i det vätskefyllda hålrummet som omger ryggmärgen i den nedre delen av ryggen. Behandlingen har dock ännu inte blivit tillgänglig för svenska patienter.
Det aktiva ämnet är en liten bit dna utformad så att den fastnar på budbärarmolekylen som cellen använder för att tillverka SOD1-genens protein. Följden blir att tillverkningen av proteinet minskar kraftigt.
Samma metod testas även mot andra muterade gener. Ett uppmärksammat fall gäller en tysk kvinna, Anna Kämpfer, som fick en ovanlig form av ALS redan som 16-åring. I hennes fall fanns mutationen i den så kallade FUS-genen. Sjukdomen utvecklades snabbt. Men efter några månaders behandling hade hon fått tillbaka tillräckligt med styrka för att själv kunna ta sig upp för en trappa. Nu pågår en klinisk studie på ett hundratal patienter i Europa och USA med mutationer i FUS-genen. Resultaten väntas bli klara under första kvartalet 2028.
Tyvärr går det inte att använda denna typ av genterapi mot alla mutationer som orsakar ALS. Det beror på att vissa mutationer finns i livsnödvändiga gener som det vore farligt att stänga av. Det fungerar inte heller i sporadiskt uppkommen sjukdom där det inte är tydligt vilken gen som ska stängas av.
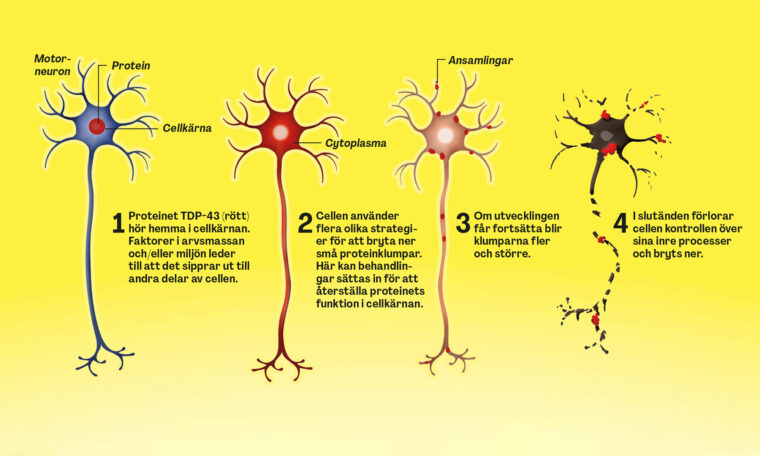
Proteinet hamnar på fel plats
Få bromsmediciner mot ALS
I dag finns bara ett fåtal mediciner som används rutinmässigt mot ALS. Den äldsta, Rilutek, kom ut på marknaden i Sverige år 1995 och förlänger livet med två till tre månader. Den tros verka genom att hämma frisättningen av signalämnet glutamat som kan skada nervceller vid för hög koncentration. Vissa patienter kan eventuellt få en bättre effekt än andra, men det är fortfarande oklart vad skillnaderna i så fall beror på.
En annan medicin, Radicava, tros fungera genom att ta hand om fria radikaler och därmed förhindra oxidativa skador i cellerna. Den kan förlänga livet med ytterligare några månader om den ges tillsammans med Rilutek.
Ett märkligt faktum om ALS är att enstaka patienter kan leva med diagnosen i decennier. Det mest kända exemplet är den brittiske fysikern och kosmologen Stephen Hawking. Han fick diagnosen som 21-åring. När han avled år 2018 hade han haft sjukdomen i 55 år.
Varför vissa patienter lever mycket längre efter diagnos än andra är i dag oklart. Det är heller inte tydligt varför patienter med nedärvd sjukdom med samma genmutation kan insjukna vid mycket olika tider i livet – det kan skilja sig med årtionden inom samma familj.
En möjlig förklaring är att påverkan av olika genmutationer blir olika stark på grund av att andra gener påverkar mutationernas effekt. Dessa så kallade sjukdomsmodifierare kan alltså försämra eller förbättra förutsättningar för olika slags motorneuroner att stå emot eller kompensera för sjukdomsförloppet.
Just nu pågår intensiv forskning om skyddande sjukdomsmodifierare, bland annat en gen som kan minska skadeverkningarna av sjukdomsgenen TDP-43. Forskning om den här typen av skyddande gener kan förhoppningsvis leda till behandlingar som inte bara hjälper patienter med specifika sjukdomsmutationer, utan mot alla former av ALS.
Texten är senast uppdaterad 21 oktober 2025 klockan 09:15.
Eva Hedlund

- Professor i neurokemi på institutionen för biokemi och biofysik vid Stockholms universitet.

Läs mer

Snart kan ALS-behandling individanpassas
Umeåforskare har skapat en analysmetod som kan göra en ny ALS-behandling mer pricksäker.

Ny genterapi bromsar ALS hos svensk patient
Forskarna vid Umeå universitet talar om ett genombrott.

Hopp om nytt sätt att behandla ALS
Ett nytt preparat har visat sig hämma ALS-sjukdomens förlamande celldöd. På sikt väcker det hopp om att kunna bromsa ett dödligt, i dag ostoppbart sjukdomsförlopp.
Är nervsignaler elektriska eller kemiska?
Vad är det egentligen som styr ryckningar i ögat?
Skolgympan ledde till en på miljonen-syndrom
Det såg ut som en stroke men var något betydligt mer ovanligt.