

Bild: Ian Haydon, UW Medicine Institute for Protein Design
AI vecklar upp proteinernas hemligheter
För proteinforskare finns det ett före och efter den 15 juli 2021. Då släpptes koden till AI-programmet Alphafold. Med en knapptryckning går det att lösa ett av vetenskapens svåraste problem: hur proteiner veckas ihop till en komplex 3D-struktur.
Demis Hassabis och John Jumper får Nobels kemipris 2024 för arbetet med Alphafold. Priset delas med David Baker.
Man kan verkligen tala om en revolution, säger Arne Elofsson, professor i bioinformatik vid Stockholms universitet.
– Det finns så klart förbättringar att göra, men generellt så är problemet med att förutsäga strukturen hos ett enskilt protein löst.
Bild: Marie Alpman
Bakom revolutionen som Arne Elofsson pratar om står ett datorprogram med artificiell intelligens, AI. Programmet har utvecklats av det brittiska företaget Deepmind som ägs av Googles ägarbolag Alphabet. Deepmind har tidigare gjort sig känt för andra AI-program, till exempel Alphago, som besegrat världsmästaren i det mycket komplicerade kinesiska brädspelet go.
Den här gången har Deepmind gett sig i kast med ett problem som forskare brottats med i 50 år, nämligen att beräkna hur nybildade proteiner veckar ihop sig till komplexa 3D-strukturer.
Utvecklingen accelererar
Efter att ha fått träna på alla redan kartlagda proteiner kan Alphafold räkna ut strukturen för ett protein vars form ännu inte analyserats med experimentella metoder. Den 15 juli 2021 valde Deepmind att publicera hur Alphafold fungerar i tidskriften Nature. Samtidigt gjordes programkoden fritt tillgänglig. Vid samma tid presenterades en utmanare, Rosettafold, som utvecklats av forskare under ledning av professor David Baker vid University of Washington, USA. Även Rosettafold är öppen för alla att använda.
Under det dryga år som gått har utvecklingen accelererat. Hundratals forskningsartiklar har skrivits som refererar till Alphafold och Rosettafold. Den vetenskapliga tidskriften Science utsåg Alphafold till årets vetenskapliga genombrott 2021.
I somras släppte Deepmind och det mellanstatliga europeiska molekylärbiologiska laboratoriet EMBL i Cambridge, Storbritannien, en databas där Alphafold beräknat strukturen hos alla kända proteiner, totalt mer än 200 miljoner. Förutom mänskliga proteiner finns strukturer för proteiner hos andra djur, växter, virus och bakterier.
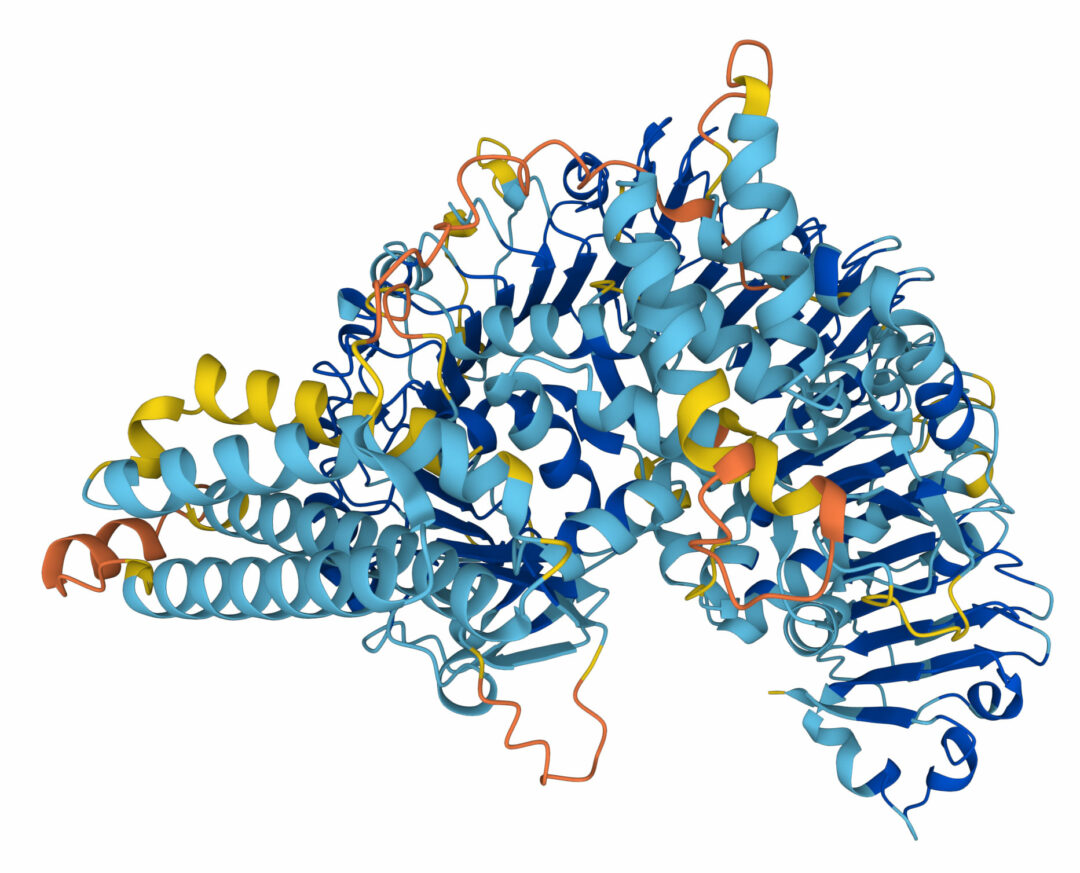
Bild: Alphafold
Proteinerna är biologins arbetshästar som sköter det mesta i kroppen. De omvandlar mat till energi, forslar syre till cellerna och får musklerna att dra ihop sig. Varje protein kodas av en gen i arvsmassan som läses av i cellernas ribosomer. Där byggs proteinerna upp i form av en kedja med 20 olika aminosyror som byggstenar. När kedjan är klar veckar den ihop sig till en komplex 3D-struktur som kan bestå av allt från några få till tusentals aminosyror. Formen är avgörande för att proteinerna ska kunna sköta sitt jobb. Förståelsen av proteinernas form är därför viktig för att förstå naturens grundläggande biologi och olika sjukdomsförlopp. Men med 20 tänkbara aminosyror på varje plats i kedjan, som alltså kan bestå av tusentals byggstenar, blir det snabbt ett astronomiskt antal olika sätt som proteinet kan ta form på. Det är detta som gör proteinveckningsproblemet så svårt att lösa.
Röntgenkristallografi och kryoelektronmikroskopi
För att kartlägga proteiners tredimensionella struktur började forskare på 1950-talet att studera hur röntgenstrålar studsar från regelbundet ordnade proteiner. Röntgenkristallografi är fortfarande en viktig metod för proteinkartläggning. Kryoelektronmikroskopi är en nyare teknik som Nobelprisbelönades 2017 och som gör det möjligt att genom snabb nedfrysning bland annat ta bilder av proteiner inne i celler. Men båda metoderna är tidskrävande och det kan ta år att kartlägga ett enda protein. Av alla de hundratals miljoner proteiner som finns i naturen var endast cirka 185 000 kartlagda och inlagda i databasen Protein data bank år 2021.
Parallellt med de här experimentella metoderna har forskare försökt lösa proteinveckningsproblemet med datorns hjälp. Ett startskott kom när den amerikanska biokemisten Christian Anfinsen tog emot sitt Nobelpris 1972. Han uttalade då visionen om att det en dag skulle bli möjligt att förutsäga 3D-strukturen hos vilket protein som helst utifrån sekvensen av aminosyror.
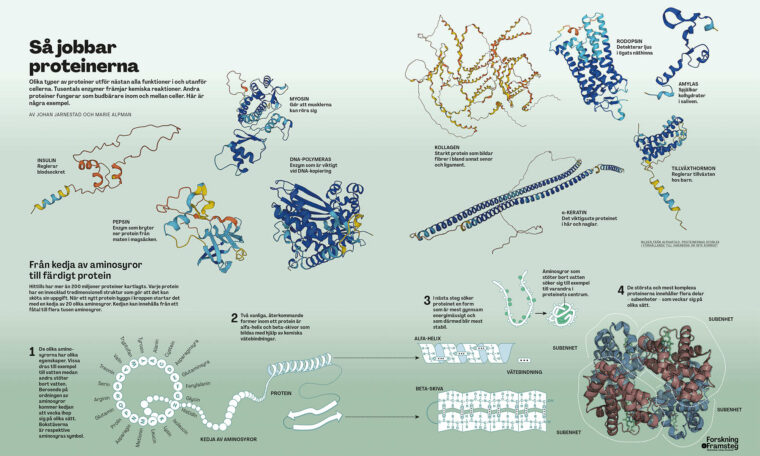
Så jobbar proteinerna
Datamodeller mäts mot varandra
I mitten av 1990-talet startade organisationen CASP en tävling där datamodeller mäts mot varandra. CASP:s tävling hålls vartannat år och går ut på att räkna ut den tredimensionella strukturen hos omkring 100 proteiner. De deltagande lagen får veta i vilken ordning aminosyrorna sitter. Utmaningen är att utifrån denna sekvens räkna ut proteinernas form. Resultaten jämförs sedan med strukturen som kartlagts experimentellt.
Med tiden har modellerna blivit allt bättre, framför allt när det gäller att förutse strukturen hos mindre proteiner. Men det var först med Alphafold som en dator blev lika bra som de experimentella metoderna. I CASP som avgjordes 2020 utklassade Alphafold de övriga lagen och kunde förutse strukturen lika bra som experimenten för fler än två tredjedelar av proteinerna.
– Det fungerar imponerande bra. En styrka hos programmet är också att det talar om när det inte vet hur strukturen ser ut, säger Arne Elofsson, som själv deltagit i samtliga CASP.
Ett exempel på vad modellerna kan användas till kom i somras. Då publicerades den första detaljerade modellen av porerna i cellkärnans membran som sköter transporten av ämnen in och ut ur cellkärnan. Det är ett enormt komplex som består av mer än 1 000 samverkande proteiner. Modellen blev möjlig tack vare en kombination av data från experiment och strukturer som beräknats av Alphafold och Rosettafold.
Forskare använder modellerna i jakten på allt från enzymer som kan bryta ner plast till nya läkemedel. Alphafold har även kommit i en version som kan modellera flera proteiner tillsammans.
Arne Elofsson och hans forskargrupp har använt den för att modellera strukturen hos proteiner som samverkar i människokroppen. I en ännu inte publicerad studie har de gått igenom alla kända proteininteraktioner, totalt 65 000.
– Av dessa kan vi få bra modeller av kanske 5 000 men många interagerar inte på ett så stabilt sätt att de går att modellera.
Alphafold bygger på djupinlärning
Alphafold bygger på så kallad djupinlärning. AI-nätverket har matats med träningsdata i form av alla kända proteiner i Protein data bank. En annan viktig input är data om hur proteiner utvecklats över tid. Jämförelser mellan hur ett protein ser ut i olika arter hjälper nätverket att förstå vilka aminosyror som är viktiga, eftersom de inte skiljer sig lika mycket åt mellan arter som andra proteiner.

Bild: Marie Alpman
– Man kan också se hur parvisa förändringar i arvsmassan hänger ihop. En förändring på en position kan innebära en annan förändring en bit bort, säger Björn Wallner, professor i bioinformatik vid Linköpings universitet som forskar om AI-modeller för att förutsäga proteiners struktur.
Han säger att det finns ett före och efter Alphafold när det gäller proteinmodellering. Men trots det har programmet begränsningar. Alphafold kan till exempel inte förutsäga formen på ovanliga proteiner som inte liknar något som det tränats för, eller förutse hur ett proteins form förändras av sjukdomsalstrande mutationer.
Bäst resultat fås för väl strukturerade proteiner, men många proteiner är oordnade och får en definierad struktur först när de samverkar med andra proteiner eller molekyler som dna, rna eller små signalämnen. Proteinet kan då anta flera olika former när det binder och sedan släpper. Men modellerna som programmet spottar ur sig är statiska.
– Livet är inte statiskt. För att utföra något behöver proteinerna röra på sig och interagera med andra proteiner, säger Björn Wallner som studerar just sådana här oordnade processer.
Ett av de proteiner han forskar om kallas Myc och har betydelse för cancer. När han ber Alphafold generera en modell av proteinet består vissa delar av tydliga och karaktäristiska spiraler. Däremellan sitter andra delar som mest liknar kokt spaghetti.
– Det är oordnade delar där Alphafold inte kan avgöra hur strukturen ser ut, förklarar han.
Genom att modifiera Alphafold försöker Björn Wallner få programmet att generera flera olika varianter av Myc för att studera hur proteinet växelverkar med andra proteiner.

Bild: Marie Alpman
Andra forskare på labb runt om i världen jobbar med att vässa algoritmerna för att ge bättre förutsägelser om hur olika proteiner interagerar. Det är viktigt om Alphafold ska kunna bli ett redskap för att utveckla nya läkemedel, något som kräver detaljerad kunskap om hur proteiner binder till andra molekyler. För det ändamålet har Deepmind skapat avknoppningsbolaget Isomorphic labs, som ska ta fram nya läkemedelskandidater med hjälp av Alphafold och andra AI-verktyg.
Kompletterande metoder
Även om Alphafold utgör ett stort steg framåt för att förutsäga proteiners struktur så löser det inte allt, konstaterar Marta Carroni, chef för avdelningen för kryoelektronmikroskopi vid Scilifelab i Stockholm.
– Det innebär inte att vi som studerar proteiner med experimentella metoder blir arbetslösa.
I stället ser hon en accelererande utveckling framför sig där experimentella data kombineras med datorgenererade modeller.
– Metoderna kompletterar varandra. Där våra bilder är suddiga kan vi ta hjälp av datormodeller.
Den växande databasen med proteinmodeller kan också fungera som vägledning och ge en vink om formen på ett nytt protein som ska undersökas.

Bild: Marie Alpman
Nya AI-verktyg används inte bara för att förutsäga proteiners form. Marta Corroni berättar att AI blivit ett viktigt redskap för att analysera den stora mängd bilder på proteiner som kommer ut ur kryoelektronmikroskopen.
Tillverkar nya proteiner
AI används också för att tillverka helt nya proteiner. Pionjären på området är David Baker, upphovsmannen till Rosettafold. Med hjälp av Alphafold, Rosettafold och andra AI-program har hans grupp visat att det går att skapa proteiner som inte finns i naturen och som skulle kunna bli nya katalysatorer i industrin eller nya antivirala läkemedel.
Ingemar André, som leder en grupp inom proteindesign vid Lunds universitet, har forskat i Bakers labb och tror att det kommer nya AI-verktyg som kan gå åt andra hållet jämfört med Alphafold. I stället för att utgå från en sekvens av aminosyror utgår de från en önskad form på ett protein och genererar sedan aminosyrafrekvensen. Därifrån är steget inte långt till att tillverka själva proteinet, till exempel i bakterier. – De här metoderna finns ännu inte men det börjar komma varianter åt det här hållet. Det är en väldigt spännande utveckling, säger Ingemar André.

Läs mer

Nobelprisad teknik avslöjar virusets minsta delar
På rekordtid har forskare kartlagt viktiga proteiner hos det nya coronaviruset – kunskap som kan ge nyckeln till ett läkemedel.
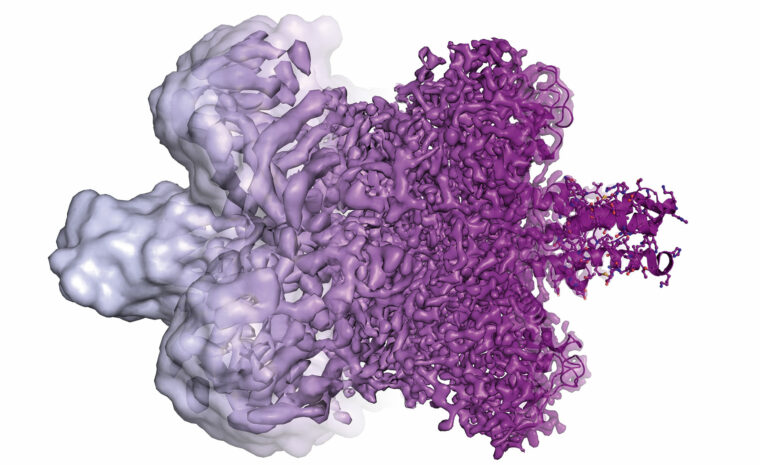
Ett kikhål in i livets maskineri
Pristagarna har gjort det möjligt att se hur enskilda molekyler arbetar inuti våra celler. Tekniken har utlöst en vetenskaplig ketchupeffekt inom framför allt den medicinska forskningen.
AI löser proteinets gåta
I mer än 50 år har försöken pågått. Nu har forskare med hjälp av AI för första gången lyckats räkna fram strukturen hos okända proteiner.

AI tar sig an Astrid Lindgrens krumelunser
Att kunna söka fritt i svårlästa gamla handskrifter är en önskedröm för släktforskare och historiker.
”AI-utvecklingens energikostnad är ohållbar”

Vi måste börja använda AI mer ändamålsenligt, skriver Adam Wickberg.



































