Alphafold klarar större proteinmodeller – tack vare svenska forskare
Det Nobelprisade AI-programmet Alphafold har fått en ny svensk uppgradering. Med verktyget AF_unmasked kan forskare i Linköping nu förutsäga stora och komplexa proteinstrukturer med högre precision. Det öppnar för viktiga framsteg inom både forskning och biomedicin.

Claudio Mirabello och Björn Wallner vid Linköpings universitet har tidigare utvecklat en föregångare till Alphafold som inspirerade Google Deepmind i utvecklingen av AI-verktyget.
Bild: Thor Balkhed
När Google Deepminds AI-program Alphafold lanserades 2020 löstes ett vetenskapligt problem som förbryllat forskare sedan 1960-talet: att exakt förutsäga hur aminosyrakedjor viker sig för att bilda proteiners tredimensionella strukturer.
Eftersom proteinernas form är avgörande för centrala biologiska funktioner som immunförsvar, cellkommunikation och muskelsammandragningar, var Alphafold ett betydande genombrott och forskarna bakom AI-programmet hedrades med Nobelpriset i kemi 2024. Men Alphafold har fortfarande betydande begränsningar.

Bild: Olov Planthaber, Linköpings universitet
– Alphafold löste problemet med att förutse vikningen av små och relativt enkla proteiner, men modellen har svårt att hantera stora proteinstrukturer där flera proteiner interagerar, säger Björn Wallner, professor i bioinformatik vid Linköpings universitet.
AF_unmasked förbättrar Alphafold
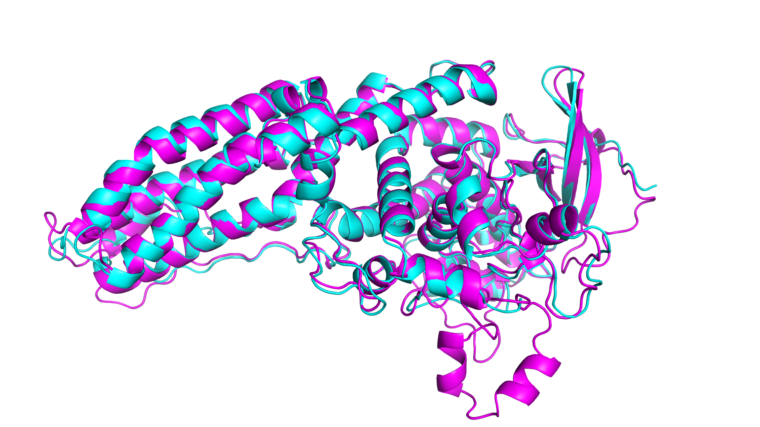
Björn Wallner och hans kollegor vid Linköpings universitet har nu utvecklat ett tillägg till Alphafold kallat AF_unmasked som övervinner flera av dessa utmaningar. Genom att kombinera data från redan kända proteinstrukturer med Alphafold kan AF-unmasked förutsäga mer komplexa proteinstrukturer med hög precision.
– Detta ger modellen en mer pålitlig och fokuserad utgångspunkt, vilket minskar sökrymden för möjliga proteinveckningar och gör AF_unmasked bättre på att hantera mer komplexa fall, säger Björn Wallner.
Passar stora proteinstrukturer
Forskarna har framgångsrikt testat verktyget på särskilt utmanande proteiner som tidigare varit svåra att modellera på grund av sin storlek och komplexitet. Genom att använda AF_unmasked har de för första gången kunnat förutsäga proteinstrukturer med upp till 10 000 aminosyror i ett enda beräkningssteg, visar studien som är publicerad i Nature Communications.
– Metoden gör det också möjligt att modellera proteiners olika tillstånd och kanske också dynamik, vilket kan ge nya insikter i hur de fungerar under olika biologiska förhållanden, berättar Björn Wallner.
”Möjligheterna för proteindesign är oändliga”
Med AF_unmasked tar forskarna ett stort steg framåt inom proteinmodellering, vilket kan få betydelse för både grundforskning och tillämpad biomedicin. Möjligheten att förutsäga och analysera stora och dynamiska proteinkomplex öppnar dörrar för nya insikter om proteinernas roll i cellfunktioner och sjukdomsprocesser.
– Det går att utveckla proteiner för användning både inuti och utanför kroppen. Möjligheterna för proteindesign är oändliga, bara fantasin sätter gränser, avslutar Björn Wallner.